Alpha-synuclein, an intrinsically disordered protein
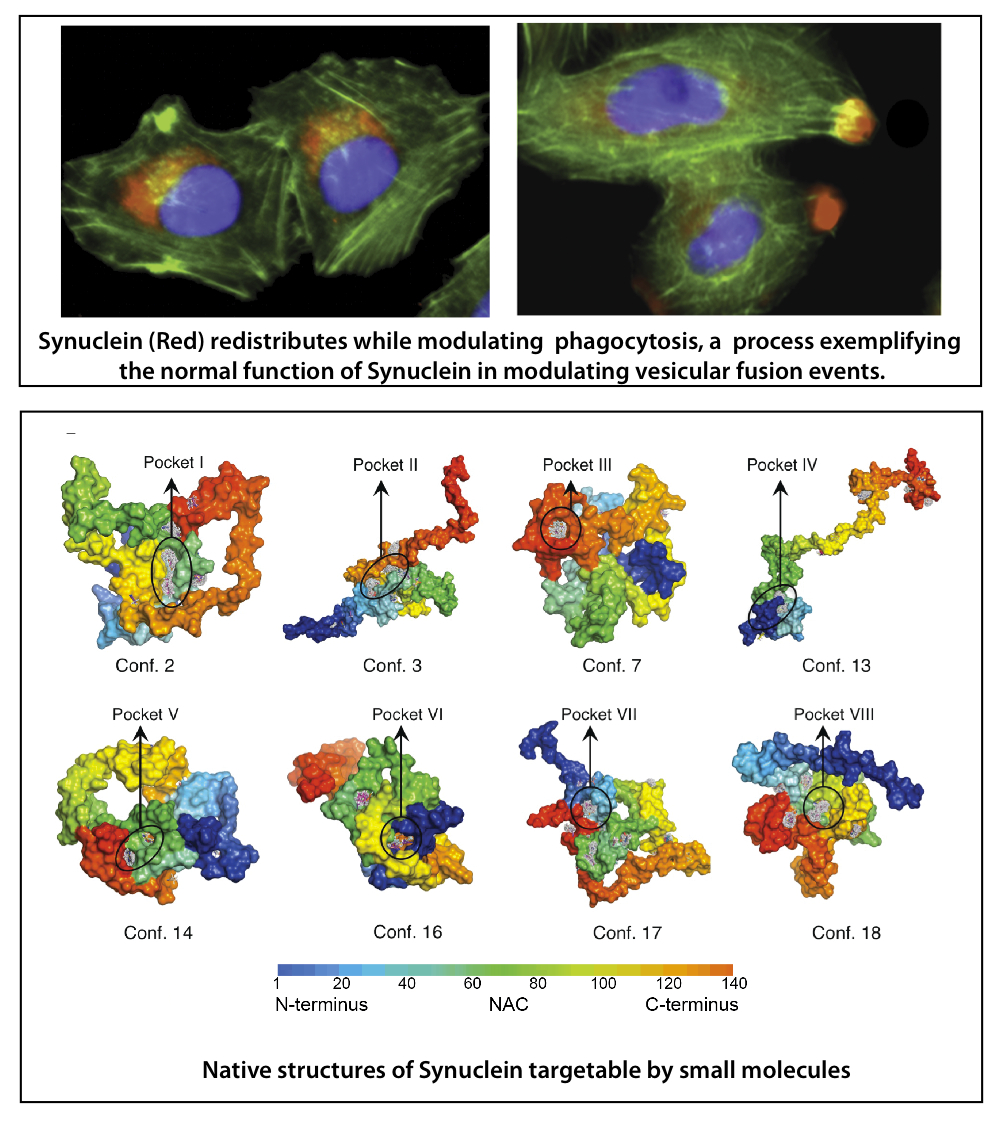
Alpha-synuclein (ASyn) was identified as a key player in Parkinson’s disease (PD) based on both human genetic and pathological evidence. Mutations or genetic multiplications of wild type protein in humans drive the development of genetically inherited forms of PD and misfolded oligomeric and fibrillar forms of ASyn are predominant features of the pathology of PD. Synuclein also plays a key role in related synucleinopathies including Alzheimer’s disease (AD) and Diffuse Lewy’ Body disease. ASyn normally functions in facilitating vesicle fusion in biological processes such as neurotransmitter release and phagocytosis. Excess ASyn, at levels that drive the development of PD in humans, leads to dysfunctional vesicular fusion events and impaired phagocytosis and neurotransmitter release. In addition, in disease tissue it forms amyloid-type toxic aggregates. Both processes are suspect in disease as there is evidence that excessive ASyn protein, such as is seen in PD and AD, can both perturb ASyn function and form toxic amyloid aggregates.
Small molecule chaperones targeting alpha-synuclein
We have taken a chemical-genetic approach to elucidate the role of ASyn malfunction and misfolding in disease and to develop therapies. We have identified small molecule chaperones that can either prevent ASyn misfolding and/or restore its normal function in modulating vesicular fusion events. We are the first to demonstrate that small molecules can target proteins with the dynamic conformational states that ASyn and other intrinsically disordered proteins have and serve as pharmacological chaperones restoring function and proper folding for this class of dynamic proteins.
To obtain these pharmacological chaperones, we identified small molecules binding to native states of ASyn in in silico and biophysical screens. Among these, we used functional assays to identify compounds blocking ASyn amyloid type aggregation and other compounds potently reversing ASyn impairment of phagocytosis under neuropathological conditions. Many amyloid forming proteins associated with neurodegenerative diseases, including ASyn, have recently been shown to spread their aggregated forms between cells and across the brain, much like prions, thereby potentially spreading disease. We are now to testing our small molecule chaperones in blocking this process and thereby identifying additional molecules with impact on the pernicious spread of aggregated ASyn.
We are using these molecules to probe mechanisms of ASyn misfolding and malfunction in disease and as leads for therapeutic drugs. Most uniquely, we are in a position to use these molecules to probe mechanisms and pathways of ASyn in modulating vesicle fusion events, including phagocytosis and neurotransmitter release, and to determine whether loss or maladaptation of those functions play a role in disease.
Engaging cellular chaperones to target alpha-synuclein
Another exciting project in our lab has emerged from a collaboration with the laboratories of Professors Gestwicki and Agard at UCSF to investigate the interaction of ASyn with the cellular Hsp70/90 chaperone machinery. In eukaryotes, the ubiquitous Hsp70 and Hsp90 molecular chaperones facilitate folding of a broad array of proteins including many associated with neurodegenerative diseases. Hsp70 and Hsp90 are key players in maintenance of protein homeoostasis, which is the balance of protein folding, trafficking and degradation to ensure proper cellular function. Hsp70 engages directly with ASyn and is protective in cellular and animal models of PD. We have demonstrated that Hsp70 interacts in a novel manner with ASyn to prevent the formation of ASyn oligomers, an early stage low order misfolded species largely thought to be the pathological driver in disease. We show that this interaction does not engage the canonical substrate binding site in Hsp70. This affords us the opportunity to enhance Hsp70’s ability to prevent ASyn misfolding without interfering with other Hsp70substrates thus providing specificity for therapeutic action and circumventing potential untoward consequences of broadly targeting Hsp70. We have established rigorous high through put screening biochemical and cellular based assays and have initiated screening for compounds with this desirable activity. We are excited about dissecting the biochemical and biological mechanisms involved in this interaction and demonstrating its relevance to disease in cellular and animal models.
Approaches
We employ a combination of drug screening and molecular biological approaches in biochemical, cellular and animal models to probe mechanism and develop therapeutic drugs. In collaboration with others at UCSF we are additionally using biophysical and molecular genetic screening techniques to identify relevant pathways and novel molecular targets.
Questions Addressed in Ongoing Studies
• Can we identify ASyn small molecule chaperones that modulate the prion-like cell-to- cell transmission of misfolded ASyn? What do they tell us about biological mechanisms of transmission of misfolding of ASyn? Can these molecules be made into therapeutic drugs?
• What do the small molecular chaperones we identified that restore normal function of ASyn tell us about the function of ASyn and its relationship to disease? Can these molecules be made into therapeutic drugs?
• How does Hsp70 mediate its protective function in cellular and animal models of Parkinson’s disease? What is the role of the non-canonical action of Hsp70 on ASyn preventing the formation of misfolded oligomers in PD? Can we target this therapeutically?
Significance
We are focused on developing and using small molecules both as research tools to understand pathobiology of ASyn and to develop drugs for neurodegenerative diseases. Human genetics and pathology indicate that ASyn is key in PD and related dementia diseases including AD. These are major causes of movement problems and dementia and because of their tight link to age present both a large and exponentially growing social challenge.
Contributions
While in industry my lab made major contributions to Alzheimer’s and Parkinson’s diseases. We drove the development of the first animal model for AD demonstrating disease-like neuropathology. Using that model, we were the first to demonstrate neurotoxicity of soluble Aβ peptide, the key diver in AD based on genetics and histopathology. This was surprising at the time as it had been thought that only the highly insoluble plaque Aβ peptide was the pathogenic species. Our lab was among the first who simultaneously cloned the rate limiting enzyme for Aβ production, BACE1 and to validate it as an AD target. BACE inhibitors are now in phase 3 clinical trials for AD.
Having made these significant contributions to AD, our group turned our attention to Parkinson’s disease. We collaborated on the first reported antibody passive vaccines for ASyn, and antibodies for ASyn are now in multiple clinical trials. Our most recent focus is to explore the role of ASyn misfolding and malfunction in PD using chaperone approaches (see above). We have developed small molecules as pharmacological chaperones that can either prevent the misfolding and/or restore normal function of ASyn. We are investigating the molecular interaction of the cellular chaperone Hsp70 with ASyn and its role in disease, and identifying small molecules that can facilitate productive action of Hsp70 on ASyn. We are using these small molecules as research tools to probe the biology of ASyn in disease models and to develop therapeutic drugs.